ОСОБЕННОСТИ ПРЕДОСТАВЛЕНИЯ ГОСУДАРСТВЕННОЙ УСЛУГИ ПО ГОСУДАРСТВЕННОЙ РЕГИСТРАЦИИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ
1Смоленский государственный университет, Факультет экономики и управления, кафедра экономики, студент 2 года обучения
Аннотация
Данная статья посвящена обзору норм отечественного законодательства, регулирующих регистрацию, перерегистрацию, фальсификацию лекарственных средств.
Ключевые слова: контроль, лекарства, регистрация лекарственных средств, регламент
Рубрика: 12.00.00 ЮРИДИЧЕСКИЕ НАУКИ
Библиографическая ссылка на статью:
Силуяненков М.Р., Скобельцинова В.В. Особенности предоставления государственной услуги по государственной регистрации лекарственных препаратов для медицинского применения // Современные научные исследования и инновации. 2019. № 6 [Электронный ресурс]. URL: https://web.snauka.ru/issues/2019/06/89827 (дата обращения: 31.07.2026).
Лекарственные средства – вещества или их комбинации, вступающие в контакт с организмом человека или животного, проникающие в органы, ткани организма человека или животного, применяемые для профилактики, диагностики (за исключением веществ или их комбинаций, не контактирующих с организмом человека или животного), лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности и полученные из крови, плазмы крови, из органов, тканей организма человека или животного, растений, минералов методами синтеза или с применением биологических технологий. [1]
К лекарственным средствам относятся фармацевтические субстанции и лекарственные препараты.
Фармацевтическая субстанция – лекарственное средство в виде одного или нескольких обладающих фармакологической активностью действующих веществ вне зависимости от природы происхождения, которое предназначено для производства, изготовления лекарственных препаратов и определяет их эффективность. [1]
Лекарственные препараты – лекарственные средства в виде лекарственных форм, применяемые для профилактики, диагностики, лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности. [1]
Давайте рассмотрим, какие лекарственные препараты могут подлежать государственной регистрации, а какие не допускаются.
Государственной регистрации подлежат следующие категории ЛП:
- лекарственные препараты, впервые подлежащие вводу в обращение в Российской Федерации;
- лекарственные препараты, зарегистрированные ранее, но произведенные в других лекарственных формах в соответствии с перечнем наименований лекарственных форм, в новой дозировке при доказательстве ее клинической значимости и эффективности;
- новые комбинации зарегистрированных ранее лекарственных препаратов. [2]
Государственной регистрации не подлежат:
- лекарственные препараты, изготовленные аптечными организациями, ветеринарными аптечными организациями, индивидуальными предпринимателями, которые имеют лицензию на фармацевтическую деятельность, по рецептам на лекарственные препараты и требованиям медицинских организаций, ветеринарных организаций;
- лекарственные препараты, приобретенные физическими лицами за пределами Российской Федерации и предназначенные для личного использования;
- лекарственные препараты, ввозимые в Российскую Федерацию для оказания медицинской помощи по жизненным показаниям конкретного пациента на основании разрешения, выданного уполномоченным федеральным органом исполнительной власти;
- лекарственные средства, ввозимые в Российскую Федерацию на основании выданного уполномоченным федеральным органом исполнительной власти разрешения и предназначенные для проведения клинических исследований лекарственных препаратов и (или) проведения экспертизы лекарственных средств для осуществления государственной регистрации лекарственных препаратов;
- фармацевтические субстанции;
- радиофармацевтические лекарственные препараты, изготовленные непосредственно в медицинских организациях в порядке, установленном уполномоченным федеральным органом исполнительной власти;
- лекарственные препараты, производимые для экспорта. [2]
Не допускается государственная регистрация:
- лекарственных препаратов, отличающихся друг от друга качественным составом действующих веществ, под одинаковым торговым наименованием;
- одного лекарственного препарата, выпускаемого производителем под различными торговыми наименованиями и представленного на государственную регистрацию в виде двух и более лекарственных препаратов. [2]
Регистрацией лекарственных препаратов и фармацевтических субстанций возложена на Министерство Здравоохранения Российской Федерации (сокр. Минздрав России) // https://www.rosminzdrav.ru
В Минздраве России данной процедурой занимается специально созданный в 2010 году Департамент государственного регулирования обращения лекарственных средств, в ведомстве которого находятся вопросы регистрации новых и обращения ранее зарегистрированных лекарственных препаратов.
Минздрав России принимает решение о регистрации лекарственных препаратов, на основании результатов проведенных экспертиз подведомственным учреждением – ФГБУ НЦЭСМП Минздрава России (Федеральное бюджетное государственное учреждение Научный центр экспертизы средств медицинского применения // www.regmed.ru).
За государственную регистрацию или внесение изменений заявитель оплачивает государственную пошлину на лицевой счет Минздрава России // величина госпошлины; реквизиты для оплаты госпошлины.
Департаментом государственного регулирования создан государственный портал // http://grls.rosminzdrav.ru для отслеживания прохождения необходимой процедуры в личном кабинете, т.к. документы для регистрации лекарственного препарата подаются в бумажном и электронном виде (оба варианта). На портале размещен Государственный реестр лекарственных средств и Государственный реестр предельных отпускных цен. Оба реестра обновляются в онлайн режиме Минздравом России и содержат самую достоверную информацию о зарегистрированных лекарственных препаратах, т.к. являются официальными государственными реестрами.
Заявителем регистрации может быть юридическое лицо, действующее в собственных интересах или юридическое лицо уполномоченное представлять интересы другого юридического лица/разработчика/производителя лекарственного препарата или фармацевтической субстанции (на основании доверенности или договора о контрактном производстве).
Регистрация лекарственных препаратов проходит несколько этапов. Поэтому давайте рассмотрим каждый этап отдельно.
Процедура регистрации зарубежных и российских препаратов идентична. Процесс регистрации лекарственного препарата состоит из 2-х основных последовательных этапов: предрегистрационные процедуры и регистрация лекарственного препарата.
Каждый этап включает несколько параллельных подпунктов:
Этап 1. Предрегистрационный этап.
П.п. 1. GMP (Надлежащая производственная практика) инспектирование производителя российскими инспекторами Минпромторга РФ (обязательно с 01.01.2016 г.)
П.п. 2. Клиническое исследования лекарственного препарата в РФ // в соответствии с ФЗ-61, статья 18, п. 10 – не все лекарственные препараты требуют клинических исследований) [3]
П.п. 3. Составление регистрационного досье по формату общего технического документа (новые требования в соответствии с // Приказом Министерства здравоохранения РФ от 12 июля 2017 г. N 409н и подготовка образцов препарата для регистрационной экспертизы. [4]
(обязательно для всех препаратов)
Этап 2. Регистрация лекарственного препарата (подпункты проводятся параллельно)
П.п. 2. Экспертиза качества лекарственного средства
П.п. 3. Экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата (польза-риск)
Из выше сказанного следует, что второй этап процедуры регистрации «регистрация лекарственного препарата» может быть начат только при наличии результатов всех процедур первого этапа.
Обязательные документы, которые должны быть в досье, чтобы не получить отказ, должны быть:
- заключение о соответствии производства зарубежного производителя правилам надлежащей производственной практики (GMP сертификат), выданное Минпромторгом РФ;
- отчет о проведенном клиническом исследовании в РФ (в случае если для данного препарата требуется проведение клинического исследования в РФ) (категории лек. препаратов для которых не нужны КИ в России Вы можете изучить, пройдя по ссылке - // ФЗ-61, статья 18, п. 10); [3]
- собрано досье по формату ОТД
- образцы одной серии препарата для предоставления на экспертизу качества (сертификат анализа на данную серию должен быть представлен в досье);
Категории лекарственных препаратов, для которых не требуется проведение клинических исследований в России, для целей государственной регистрации // ФЗ-61, статья 18, п. 10:
- Если препарат, заявленный на регистрацию, является воспроизведенным (дженериком), а референтный ему препарат зарегистрирован и применяется в России более 20 лет.
- Если проведены международные многоцентровые исследования с включением России.
- Если препарат с идентичным составом референтному препарату, водным раствором для парентерального (подкожного, внутримышечного, внутривенного, внутриглазного, внутриполостного, внутрисуставного, внутрикоронарного) введения.
- Если препарат с идентичным составом референтному препарату, раствором для перорального применения.
- Если препарат с идентичным составом референтному препарату, и произведен в форме порошка или лиофилизата для приготовления растворов.
- Медицинские газы
- Если препарат с идентичным составом референтному препарату, ушным или глазным лекарственным препаратом, произведенным в форме водного раствора.
- Если препарат с идентичным составом референтному препарату, водным раствором для местного применения.
- Если препарат с идентичным составом референтному препарату и представляют собой водный раствор для использования в форме ингаляций с помощью небулайзера или в качестве назального спрея, применяемого с помощью сходных устройств. [3]
Второй этап процесса регистрации «регистрация лекарственного препарата»:
Регистрационное досье собранное по формату ОТД подается в Минздрав России. В течение 10 рабочих дней проводится проверка комплектности досье и принимается решение о направлении документов на экспертизу:
- Проводится только для орфанных препаратов! экспертизу документов, представленных для определения возможности рассматривать лекарственный препарат для медицинского применения при осуществлении государственной регистрации в качестве орфанного лекарственного препарата;
- экспертизу заявленных в нормативной документации методов контроля качества лекарственного средства и качества представленных образцов лекарственного средства с использованием этих методов (далее – экспертиза качества);
- экспертизу отношения ожидаемой пользы к возможному риску применения лекарственного препарата.
По результатам проведенных экспертиз оформляется экспертное заключение или запрос о недостаточности представленных данных и передается из экспертного учреждения (ФГБУ «НЦЭСМП») в Минздрав России.
В случае запроса, Заявитель должен в течение 90 рабочих дней исправить полученные замечания и направить из Минздрав, они в свою очередь направляют документы в экспертную организацию. Запросы могут быть повторными, если Заявитель не в полной мере ответил на поставленные вопросы.
При положительном заключении (после устранения замечаний или при их отсутствии) эксперты Минздрава России в течение 10 рабочих дней направляю проект Регистрационного удостоверения (РУ) на согласование Заявителю по электронной почте, указанной в личном кабинете, после согласования РУ вносят препарат в государственный реестр лекарственных средств и оформляют РУ. Если в результате проведенной экспертизы невозможно подтвердить качество или эффективность/безопасность препарата, тогда выдается решение об отказе в гос. регистрации препарата.
При первой регистрации препарата в России РУ выдается на 5 лет. По истечении данного срока производитель подает документы для подтверждения регистрации препарата и тогда уже регистрационное удостоверение выдается бессрочно.
Ниже приведено наглядное пособие всего того, о чем было сказано ранее. Данная блок схема поможет понять суть регистрации лекарственных препаратов.
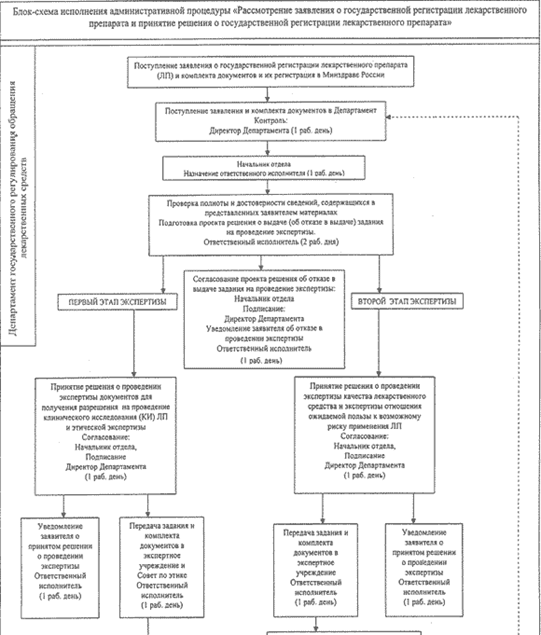
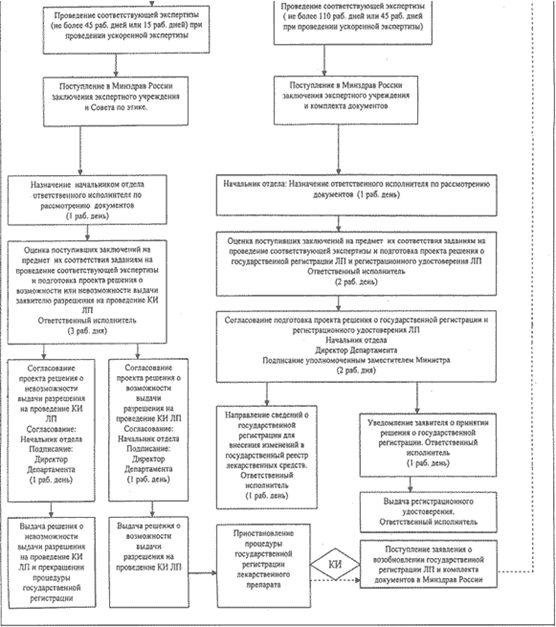
Таким образом, данный регламент помогает людям в их проблемах с регистрацией, перерегистрацией и борьбой с фальсификацией лекарственных средств. Объясняет каким способом проходит проверка лекарственных препаратов и каким способом зарегистрировать их. Рассказывает о необходимых документах для выпуска лекарственных препаратов и сроках их подтверждения. И не удивительно что медикаменты проходят такую тщательную проверку. Ведь здоровье каждого человека очень важно и не должно страдать он некачественных препаратов.
Библиографический список
- ФЗ от 12.04.2010 №61-ФЗ (ред. от 27.12.2018) Статья 4. Основные понятия, используемые в настоящем Федеральном законе (Электронный ресурс) URL: // http://www.consultant.ru/document/cons_doc_LAW_99350/baabe5b69a3c031bfb8d485891bf8077d6809a94/ (дата обращения 10.06.2019)
- ФЗ от 12.04.2010 №61-ФЗ (ред. от 27.12.2018) Статья 13. Государственная регистрация лекарственных препаратов (в ред. Федерального закона от 22.12.2014 N 429-ФЗ (Электронный ресурс) URL: // http://www.consultant.ru/document/cons_doc_LAW_99350/d51604f907a67575d6f37a17e2322f91bee26db8/ (дата обращения 10.06.2019)
- ФЗ от 12.04.2010 №61-ФЗ (ред. от 27.12.2018) Статья 18. Подача и рассмотрение заявления о государственной регистрации лекарственного препарата для медицинского применения (Электронный ресурс) URL: // http://www.consultant.ru/document/cons_doc_LAW_99350/6d9c982585a1928071e3b1c9897ab4d2ec97e182/ (дата обращения 10.06.2019)
- Электронный ресурс. URL: // http://www.consultant.ru/document/cons_doc_LAW_222249/ (дата обращения 10.06.2019)
Все статьи автора «Силуяненков Михаил Романович»
© Если вы обнаружили нарушение авторских или смежных прав, пожалуйста, незамедлительно сообщите нам об этом по электронной почте.